Lymphangioleiomyomatosis (LAM) is a rare lung disease found primarily in women and often affects other organs including lymph nodes and kidneys. LAM is caused by inactivating mutations in the tuberous sclerosis complex (TSC) genes (TSC1 and TSC2), resulting in activation of the mammalian target of rapamycin (mTOR) complex 1 (TORC1) signaling network. LAM can occur in otherwise healthy women (sporadic LAM) or in women who have TSC, a genetic disease associated with benign tumors of the brain, heart, skin, and kidneys. Hyperactivation of TORC1 in LAM cells leads to growth factor-independent growth, increased cell size, enhanced cell survival and suppressed autophagy. Although their origin is not known, LAM cells resemble smooth muscle cells, express melanocyte-lineage proteins, and ultimately metastasize to the lungs. The metastasis to lungs is believed to occur via both lymphatic and blood circulation, resulting in cystic lung destruction.
Symptoms
The initial symptoms of LAM can include shortness of breath, coughing of blood (hemoptysis), spontaneous bleeding from a renal angiomyolipoma, or spontaneous lung collapse (pneumothorax). LAM can be very difficult to diagnose since at its earlier stages, there are usually no distinctive signs apparent on physical examination or chest X-ray. Over time, LAM can cause cystic destruction of the lung that eventually leads to respiratory failure (see Figure below). The average age at diagnosis is 35 and after 10 years approximately 55% of LAM patients experience shortness of breath, 20% require supplemental oxygen, and 10% have died.
Normal Lungs and Lungs With LAM
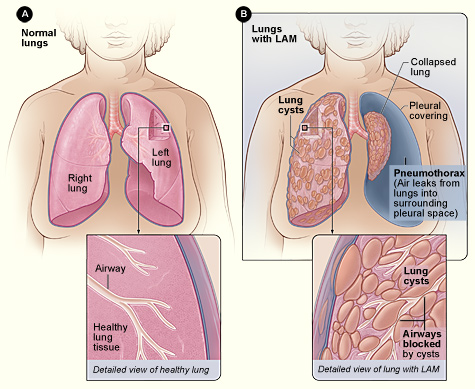
Figure A shows the location of the lungs and airways in the body. The inset image shows a cross-section of a healthy lung. Figure B shows a view of the lungs with LAM and a collapsed lung (pneumothorax). The inset image shows a cross-section of a lung with LAM. For more information please visit the National Institutes of Health.
Incidence
There are at least 200 new cases of LAM diagnosed each year in the United States. LAM affects every race and has been found in women in over 60 countries. LAM is often undiagnosed or misdiagnosed and women may have symptoms for years before a correct diagnosis is made.
Treatment
Sirolimus/rapamycin is the only approved drug for LAM. A Phase 3 trial has shown that sirolimus stabilizes lung function in moderate to severely affected women with LAM. However, sirolimus has side effects and after cessation of treatment, lung function decline resumed. Other treatments for LAM include diuretics, bronchodilators, oxygen supplementation and lung transplantation.
For more resources about Lymphangioleiomyomatosis, please see LAM Foundation and LAM Health Project.